Results and Conclusions#
Interpretation of the Results#

First, we visually check the structure. This is the last snapshot of the simulation, with the box replicated for a better view. The C–S–H layers are preserved, the pore space has closed, and overall we can observe that the structure is physically sound
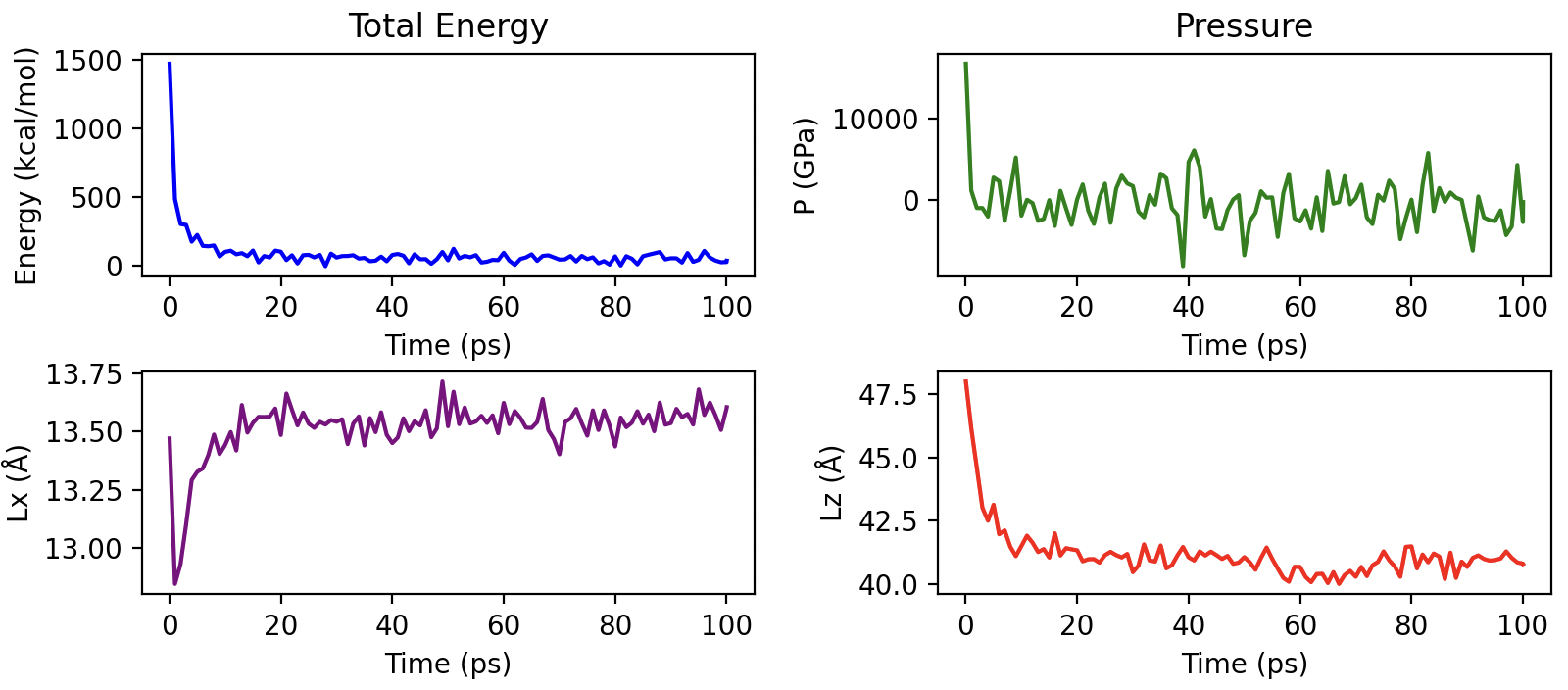
Then we analyze several parameters to check convergence. The total energy decreases very fast after the initial relaxation of the atomic positions. It seems that the system has converged, but we recommend to zoom in to guarantee convergence. Same interpretation could be done with the pressure. Regarding lattice parameters, the x dimension presents a strong fluctuation in the very beginning, but the system recovers the initial value after 20ps. The changes in the z dimension are much more pronounced as the pore decreases its size to equilibrate the water density inside the pore.
The density profile of Cl ions and water in your system should look like this:
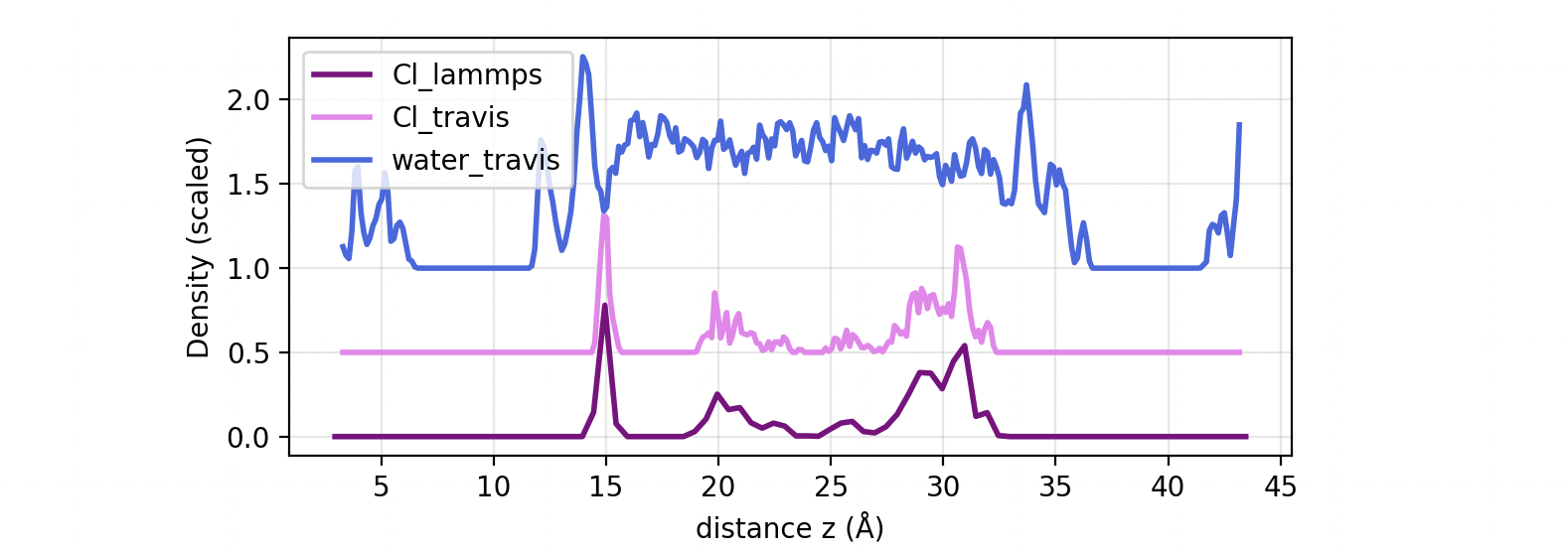
The results for Cl computed from LAMMPS and TRAVIS are equal, with the only difference of the bin resolution, 0.5 in LAMMPS and \(\approx\) 0.13 in TRAVIS depending on your settings. The water dprof shows 2 peaks of well structured water close to the surfaces (clearer in the bottom) followed by “bulk” water in the center of the pore. Cl ions are located preferentially at the surfaces. In the bottom surface, the structure is clearer, with Cl ion(s) in a fixed site just after the 2nd water layer.
The MSD of Cl ions in the pore looks like this:
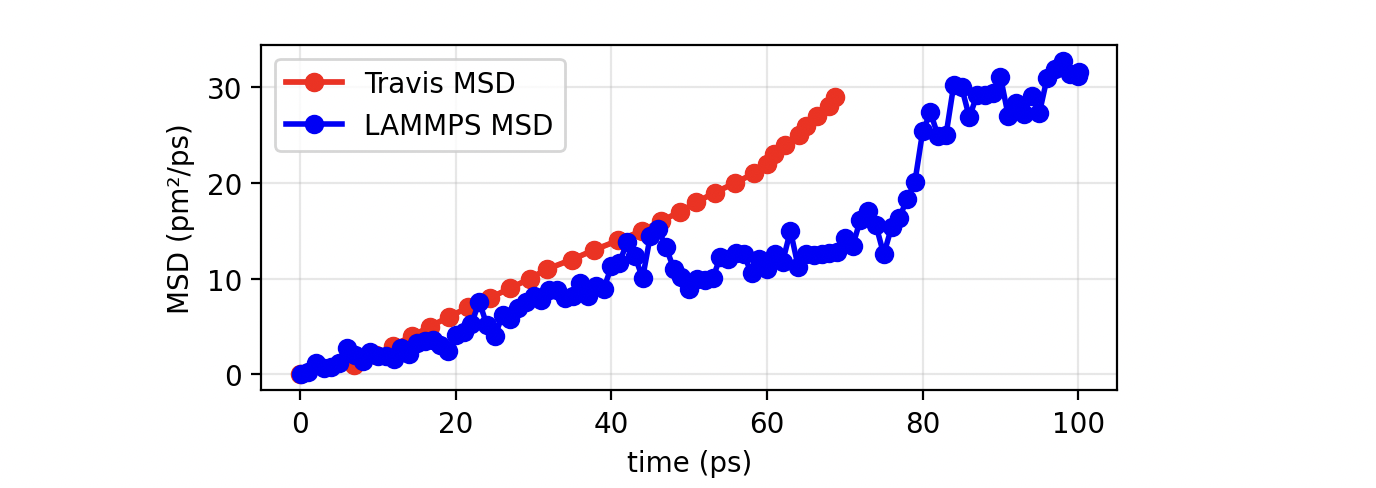
In this case, the results from TRAVIS and LAMMPS do not match perfectly, just because the algorithms used are different. LAMMPS computes the MSD on-the-fly during the simulation with the displacement always measured with respect to the initial configuration:
Only a single time origin (t₀ = 0) is used, so no temporal averaging is performed. TRAVIS calculates the MSD in post-processing from the full trajectory, averaging over many time origins:
The program uses all possible starting points times up to a defined correlation depth. This multi-time averaging provides smoother and statistically more reliable results. However, it requires saving and processing the full trajectory. Therefore, LAMMPS provides a quick estimate of diffusion and atomic displacement, while TRAVIS offers a more accurate and statistically averaged MSD, better suited for extracting reliable diffusion coefficients.
Overall Conclusions of the Course#
In this tutorial, we have shown the full workflow of atomistic simulation applied to cementitious materials: (i) building a structural model of C–S–H with a slit pore, (ii) preparing and running a molecular dynamics simulation with LAMMPS, and (iii) analyzing the output to extract physically meaningful properties such as diffusion constants and density distributions.
It is important to stress, however, that the examples presented here are highly simplified. The simulation times are much shorter than those required to obtain converged transport properties, and the models do not yet account for the full structural complexity of real C–S–H. In a research-level study, one would need to run trajectories that are hundreds of times longer, carefully test the force field and validate the results against experimental data.
Despite these limitations, the course provides an illustrative case study that demonstrates the power and limitations of molecular dynamics for cement science. Students leave the course with a clear view of how atomistic simulations are set up, run, and analyzed, and what steps are necessary to transition from a didactic example to a robust scientific study.
Future courses#
The material from this course[1] will remain permanently available. If you use it, you can cite it as doi. New material will be created for future ICASCM courses, tentatively scheduled for 2027. In addition, the UPV/EHU will offer a 25-hour course on atomistic simulation of cementitious materials in October 2026. The course will be hybrid (online and in-person), with theory and hands-on exercises. Seats are limited, you can send your expression of interest by mail to eduardo.duque@ehu.eus.